La maladie de Hunter, aussi connue sous le nom de mucopolysaccharidose de type II (MPS II), est une pathologie génétique rare qui suscite encore de nombreuses interrogations. Touchant principalement les garçons, cette affection se caractérise par un déficit enzymatique entraînant une accumulation progressive de substances complexes au sein des cellules corporelles. Cette surcharge impacte plusieurs organes et systèmes, provoquant des symptômes variés et une évolution souvent complexe de la maladie. En 2026, les avancées médicales dans la prise en charge et le traitement offrent de nouvelles perspectives pour les patients et leurs familles, malgré les défis persistent liés au caractère multisystémique de la maladie.
Le syndrome de Hunter illustre parfaitement la complexité des maladies lysosomales rares, où une enzyme défaillante perturbe le fonctionnement normal des cellules. Les symptômes affectent aussi bien l’appareil locomoteur que les fonctions cognitives, avec une progression plus ou moins rapide selon la forme de la maladie. Les progrès dans les technologies de diagnostic, couplés à une meilleure compréhension des mécanismes génétiques, permettent aujourd’hui un dépistage plus précoce et un suivi thérapeutique adapté. Comprendre les causes, reconnaître les manifestations cliniques et adopter une prise en charge individualisée sont autant d’enjeux essentiels pour améliorer la qualité de vie des enfants concernés et alléger le poids familial.
- Population touchée : maladie rare affectant majoritairement les garçons (1 naissance sur 100 000 en France)
- Origine génétique : mutation sur le chromosome X impliquant le déficit de l’enzyme iduronate-2-sulfatase
- Symptômes variés : dysmorphie faciale, troubles neurologiques, atteintes cardiaques et respiratoires
- Diagnostic : analyse enzymatique et génétique combinées pour confirmation
- Traitements : enzymothérapie substitutive, prise en charge multidisciplinaire et avancées récentes en thérapie génique
Comprendre les causes et mécanismes de la maladie de Hunter
La maladie de Hunter est avant tout une pathologie génétique due à une mutation du gène IDS situé sur le chromosome X. Cette anomalie engendre un déficit enzymatique en iduronate-2-sulfatase, une enzyme indispensable à la dégradation des glycosaminoglycanes (GAG), des sucres complexes présents dans les tissus. En raison de l’absence ou de la faible activité de cette enzyme, les GAG s’accumulent à l’intérieur des lysosomes, ces organites cellulaires chargés de recycler les déchets. Cette surcharge lysosomale altère la fonction cellulaire normale, provoquant un dysfonctionnement progressif des organes concernés.
Ce mécanisme physiopathologique explique pourquoi la maladie est progressive et multisystémique. Les lysosomes en surcharge perturbent particulièrement les tissus conjonctifs, ce qui entraîne des anomalies squelettiques, ainsi que les organes tels que le foie, la rate, le cœur et le cerveau. La transmission récessive liée à l’X implique que seuls les garçons développent la maladie, tandis que les filles sont généralement porteuses asymptomatiques. Toutefois, environ 30 % des cas correspondent à des mutations de novo, apparaissant spontanément sans antécédents familiaux.
La mucopolysaccharidose de type II peut se présenter sous deux formes principales. La forme sévère, dite neuronopathique, se manifeste par un déclin cognitif rapide, des troubles du comportement et une espérance de vie raccourcie. La forme atténuée, non-neuronopathique, se caractérise par une atteinte moins marquée du système nerveux, avec un pronostic plus favorable. Ces distinctions reflètent la diversité des mutations et de l’activité enzymatique résiduelle entre les patients.
En termes d’épidémiologie, la maladie est très rare, avec environ 1 garçon affecté sur 100 000 naissances masculines en France. L’incidence varie selon les régions, plus fréquente notamment en Asie. Malgré sa rareté, le registre français des mucopolysaccharidoses recense plus de 400 cas de MPS II, contribuant à une meilleure connaissance de son évolution et à l’optimisation des stratégies thérapeutiques.
Pour appréhender pleinement cette maladie, il est utile de consulter des ressources détaillées comme cette page encyclopédique qui expose en profondeur ses aspects médicaux et biologiques.
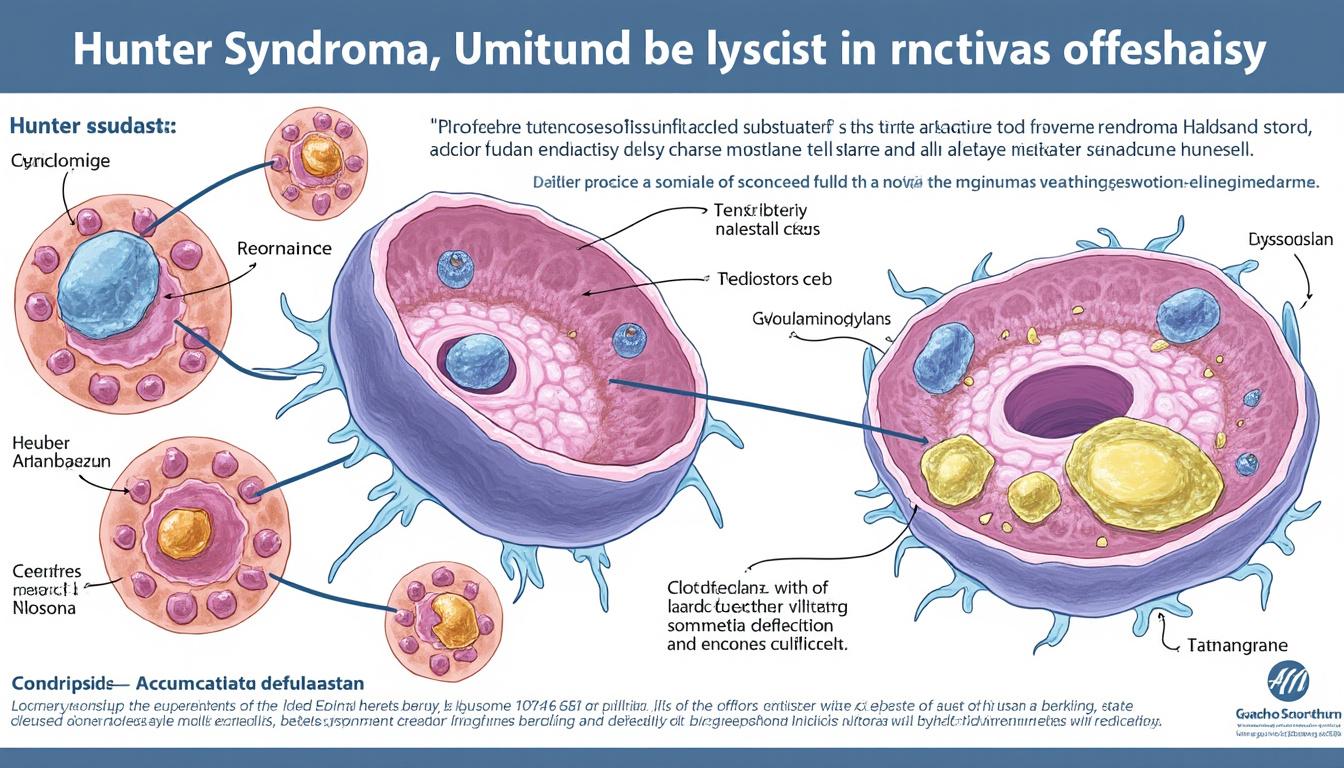
Les symptômes variés et manifestations cliniques de la maladie de Hunter
Les symptômes de la maladie de Hunter apparaissent généralement entre 2 et 4 ans, bien que certains signes puissent se manifester plus tôt ou plus tard selon la sévérité. Ils sont très divers, révélant la nature multisystémique de la mucopolysaccharidose de type II. Ces manifestations cliniques affectent principalement la morphologie, le système neurologique, le système respiratoire, et le système cardiaque.
Cliniquement, on observe un faciès caractéristique avec une dysmorphie progressive : un nez élargi, des lèvres épaissies, une langue volumineuse et une mâchoire proéminente. Squelettiquement, la maladie provoque une dysostose multiple, une atteinte osseuse entraînant raideurs articulaires, déformations osseuses et une taille basse. Les mains peuvent présenter des doigts courts, épais, ayant un aspect en griffe. Ces anomalies peuvent limiter considérablement la mobilité et la qualité de vie.
Concernant l’atteinte neurologique, elle dépend de la forme de la maladie. Les formes sévères se traduisent par un retard global du développement, des troubles du comportement et une régression des acquis moteurs et cognitifs, rendant le suivi plus complexe. Dans les formes atténuées, ces symptômes neurologiques sont nettement atténués, ce qui permet un développement intellectuel mieux préservé.
Sur le plan cardiorespiratoire, les patients présentent fréquemment des valvulopathies, une cardiomyopathie et des troubles du rythme. Les complications respiratoires sont principalement liées à une obstruction des voies aériennes supérieures, ce qui entraîne des difficultés respiratoires et des troubles du sommeil, parfois sévères. Une trachéotomie peut être nécessaire dans certains cas. Ces aspects exigent un suivi médical régulier et rigoureux pour prévenir les crises aiguës.
Les manifestations cutanées et les troubles auditifs sont également observés, bien que moins spécifiques. L’hépatosplénomégalie (augmentation du volume du foie et de la rate) est fréquente mais souvent asymptomatique au début.
Pour une description exhaustive des symptômes et de l’évolution, gesundMD propose une analyse complète.
Procédures diagnostiques essentielles pour la mucopolysaccharidose de type II
Le diagnostic de la maladie de Hunter repose sur une série d’examens spécifiques, étant donné la complexité et la rareté de la pathologie. Le premier signe qui oriente vers cette maladie est souvent la suspicion clinique basée sur l’observation des symptômes caractéristiques, suivie d’examens biologiques approfondis.
Le dépistage initial inclut le dosage des glycosaminoglycanes dans les urines, qui révèle une augmentation anormale des GAG. Cependant, ce test n’est pas spécifique à la MPS II, ce qui impose de poursuivre les investigations. La confirmation diagnostique repose sur la mesure de l’activité enzymatique de l’iduronate-2-sulfatase, réalisée sur des prélèvements sanguins ou sur des fibroblastes issus d’une biopsie cutanée. Une activité faible ou absente confirme le diagnostic.
Par ailleurs, l’analyse génétique est indispensable pour identifier la mutation responsable et réaliser un conseil génétique familial. Celle-ci est réalisée dans des laboratoires spécialisés agréés, et permet également une prise en charge anticipée des membres à risque. Le diagnostic prénatal est proposé aux couples porteurs, généralement par biopsie des villosités choriales ou amniocentèse dès 10 à 12 semaines de grossesse.
Un tableau récapitulatif des examens diagnostiques est utile pour visualiser ce parcours :
| Type d’examen | Description | Importance |
|---|---|---|
| Dosage des glycosaminoglycanes urinaires | Mesure des GAG dans les urines | Dépistage initial, non spécifique |
| Activité enzymatique | Évaluation de la fonction de l’iduronate-2-sulfatase | Diagnostic de confirmation |
| Analyse génétique | Identification de la mutation IDS | Conseil génétique et dépistage familial |
| Diagnostic prénatal | Biopsie villosités choriales ou amniocentèse | Orientation des décisions familiales à risque |
La complexité de cette maladie impose un suivi médical spécialisé et pluridisciplinaire. La téléconsultation peut permettre certaines évaluations à distance, notamment le suivi des symptômes cognitifs ou la coordination entre spécialistes, mais les consultations en présentiel restent indispensables pour le bilan clinique, les examens cardiaques, pulmonaires et neurologiques approfondis.
Les traitements actuels et la prise en charge multidisciplinaire de la maladie de Hunter
Le traitement de la mucopolysaccharidose de type II repose principalement sur la thérapie enzymatique substitutive par l’idursulfase, commercialisée sous le nom d’Elaprase. Cette enzymothérapie consiste en des perfusions intraveineuses hebdomadaires destinées à compenser le déficit en enzyme et à réduire l’accumulation des glycosaminoglycanes dans les tissus.
Les bénéfices de cette thérapie sont bien documentés : amélioration des capacités respiratoires, stabilisation de la fonction cardiaque, diminution du volume hépatique et splénique. Toutefois, son efficacité est limitée par l’impossibilité à franchir la barrière hémato-encéphalique, ce qui ne permet pas d’agir efficacement sur les symptômes neurologiques des formes sévères.
Pour ces dernières, la greffe de cellules souches hématopoïétiques peut être envisagée, notamment si réalisée précocement, avant l’âge de 2 ans. Ce traitement agressif vise à limiter les atteintes neuronales en apportant des cellules capables de produire l’enzyme déficitaire. En parallèle, la prise en charge intègre une multitude d’interventions : kinésithérapie, orthophonie, suivi cardiologique et pneumologique, soutien psychologique, ainsi qu’un accompagnement éducatif adapté.
En 2024 et 2025, les recherches ont donné lieu à plusieurs innovations thérapeutiques majeures. La thérapie génique apparaît prometteuse avec des essais cliniques en cours, et un traitement par voie intrathécale (idursulfase-IT) permet désormais une meilleure efficacité sur le système nerveux central. Par ailleurs, la mise à disposition d’enzymes à longue durée d’action réduit la fréquence des perfusions, améliorant la qualité de vie des patients.
Ces nouveautés, associées à un suivi strict selon les recommandations officielles comme celles de la Haute Autorité de Santé, illustrent une prise en charge moderne et dynamique, centrée sur le bien-être global du patient.
Un accompagnement des familles est également essentiel, avec des associations dédiées telles que VML – Vaincre les Maladies Lysosomales, spécialisées dans l’information, le soutien psychologique et l’aide à la gestion des aides sociales.

Adaptations quotidiennes et gestion du long terme chez les patients atteints de la mucopolysaccharidose de type II
Vivre avec une maladie comme la mucopolysaccharidose de type II implique des ajustements constants dans la vie familiale, scolaire et sociale de l’enfant atteint. La lourdeur du traitement, notamment les perfusions hebdomadaires qui peuvent durer plusieurs heures, nécessite une organisation rigoureuse et un soutien psychologique adapté.
À l’école, la mise en place d’un projet d’accueil individualisé (PAI) permet d’ajuster le rythme et les conditions d’apprentissage en fonction des capacités et des besoins de l’enfant. L’intervention d’un personnel formé et la communication étroite avec les équipes soignantes facilitent cette adaptation. Les aides matérielles et financières telles que la reconnaissance en affection de longue durée (ALD) et la prestation de compensation du handicap (PCH) sont des ressources incontournables pour alléger les contraintes.
L’activité physique, bien que limitée par les atteintes articulaires, reste bénéfique. Les sports doux comme la natation ou la marche contribuent au maintien de la mobilité et au bien-être général. Parallèlement, un accompagnement kinésithérapique est indispensable pour prévenir l’aggravation des déformations osseuses et articulaires.
La vie sociale et familiale est souvent mise à l’épreuve, avec des impacts psychologiques importants. L’information de l’entourage proche est fondamentale pour favoriser la compréhension et le soutien. Des réseaux d’entraide, conférences et groupes de parole proposés par les associations spécialisées sont d’un grand secours pour rompre l’isolement.
- Planification rigoureuse des rendez-vous médicaux et traitements
- Mise en place d’un PAI et suivi scolaire personnalisé
- Pratique régulière d’activités physiques adaptées
- Mobilisation des aides financières et matérielles
- Soutien psychologique et communication au sein de la famille
Ces stratégies permettent de mieux appréhender le quotidien avec la maladie de Hunter et d’améliorer sensiblement la qualité de vie sur le long terme, tout en anticipant le suivi médical nécessaire.
Quelles sont les causes principales de la maladie de Hunter ?
La maladie est causée par une mutation génétique sur le chromosome X, entraînant un déficit en iduronate-2-sulfatase, une enzyme essentielle pour dégrader certains sucres complexes, ce qui provoque une accumulation toxique dans les cellules.
Quels sont les symptômes les plus fréquents ?
Les symptômes incluent une dysmorphie faciale (nez et lèvres épaissis), des difficultés respiratoires, une atteinte neurologique variable, des déformations osseuses et des problèmes cardiaques.
Comment est posé le diagnostic ?
Le diagnostic repose sur la mesure enzymatique de l’activité iduronate-2-sulfatase et la confirmation par analyse génétique. Le dosage des glycosaminoglycanes urinaires permet un dépistage initial.
Quels traitements sont disponibles ?
La thérapie enzymatique substitutive est le traitement de référence. Pour les formes sévères, la greffe de cellules souches hématopoïétiques peut être envisagée, en complément des soins multidisciplinaires.
Comment améliorer la qualité de vie au quotidien ?
Il est essentiel d’organiser les soins, d’adapter la scolarité via un PAI, de pratiquer une activité physique adaptée et de bénéficier d’un soutien psychologique ainsi que de l’accompagnement des associations spécialisées.
